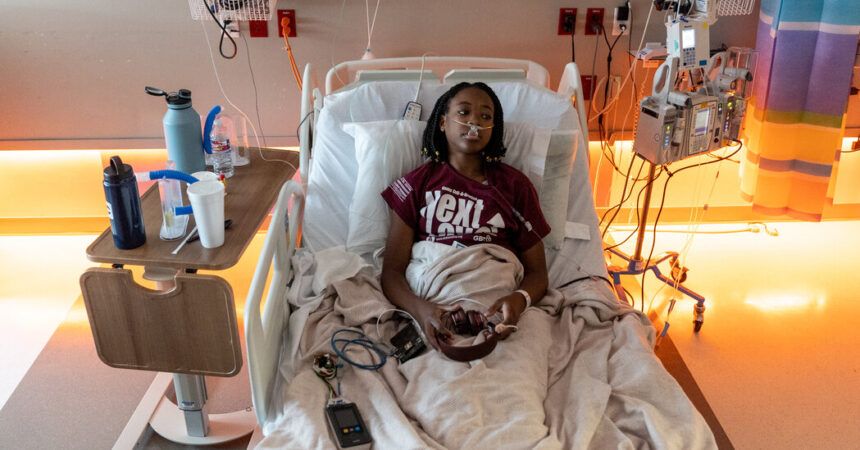
[ad_1]
Ein Expertengremium sagte am Dienstag, dass eine bahnbrechende Behandlung der Sichelzellenanämie sicher genug für den klinischen Einsatz sei und bereitete damit die Voraussetzungen für die voraussichtliche bundesstaatliche Zulassung eines wirksamen potenziellen Heilmittels für eine Krankheit, an der mehr als 100.000 Amerikaner leiden, bis zum 8. Dezember.
Die Food and Drug Administration hatte zuvor festgestellt, dass die als Exa-Cel bekannte und gemeinsam von Vertex Pharmaceuticals aus Boston und CRISPR Therapeutics aus der Schweiz entwickelte Behandlung wirksam war. Aufgrund der Schlussfolgerung des Gremiums vom Dienstag über die Sicherheit von Exa-Cel wird es an die FDA weitergeleitet, um über die Freigabe für den breiten Einsatz bei Patienten zu entscheiden.
Exa-cel befreit Patienten von den schwächenden und schmerzhaften Auswirkungen dieser chronischen, tödlichen Krankheit. Im Falle einer Zulassung wäre das Vertex-Produkt das erste Medikament, das eine genetisch bedingte Krankheit mit der Gen-Editing-Technik CRISPR behandelt.
Es könnte auch die erste einer Reihe neuer Optionen zur Heilung der quälenden Krankheit sein. Bis zum 20. Dezember wird die FDA über ein zweites mögliches Heilmittel gegen Sichelzellenanämie entscheiden, eine Gentherapie, die von der Firma Bluebird Bio aus Somerville, Massachusetts, entwickelt wurde.
Die Sichelzellenanämie wird durch eine Genmutation verursacht, die dazu führt, dass sich die Blutzellen verformen, sodass sie Sicheln oder Halbmonden ähneln. Es betrifft Millionen von Menschen weltweit, von denen die meisten afrikanischer Abstammung sind. Die deformierten Zellen bleiben in den Blutgefäßen stecken und verursachen Schlaganfälle, Organschäden und Episoden quälender Schmerzen, da den Muskeln Sauerstoff fehlt.
Der Tribut an Sichelzellenanämie beginnt schon früh im Leben. Evelyn Islam aus Milwaukee, jetzt 8 Jahre alt, hatte 22 Bluttransfusionen und musste sich die Milz entfernen lassen, bevor sie 3 Jahre alt war. „Gentherapie ist unsere letzte Hoffnung auf Heilung“, sagte ihre Mutter, Melissa Nicole Allen.
Doch für viele kommen die neuen Gentherapien zu spät.
Ashley Valentine, Mitbegründerin der landesweiten Interessenvertretung Sick Cells, musste zustimmen drei Monate frei von der Arbeit im Jahr 2016, um ihrem Bruder Marqus bei der Bewältigung der Sichelzellensymptome zu helfen. Als er 2018 einen Hüftgelenksersatz erhielt, akzeptierte ihr Vater schließlich eine Entlassung von seinem Job, um sich um ihn kümmern zu können.
„Und das sind nur wir“, sagte sie.
Marqus starb 2020 im Alter von 36 Jahren an einem durch Sichelzellenanämie verursachten Schlaganfall.
Neue Behandlungen wie die, die am Dienstag genehmigt wurde, werden voraussichtlich Millionen von Dollar pro Patient kosten, obwohl Vertex noch nicht gesagt hat, wie hoch die Kosten sein werden. Doch auch die lebenslange Betreuung der erkrankten Patienten ist enorm teuer und verursacht schätzungsweise hohe Kosten für das Gesundheitssystem 3 Milliarden Dollar pro Jahr.
Es ist noch nicht klar, wie viele Menschen die neue Therapie in Anspruch nehmen werden. Die neuen Therapien sind zudem nicht einfach zu ertragen und bringen Belastungen für die Patienten mit sich, die sich einer Chemotherapie unterziehen und mehr als einen Monat im Krankenhaus verbringen müssen. Auch Familienmitglieder sind betroffen – sie müssen sich während der intensivsten Phase der Behandlung möglicherweise eine Auszeit von der Arbeit nehmen.
Darüber hinaus sind die meisten Amerikaner mit Sichelzellenanämie Schwarze und vertrauen einem möglicherweise nicht Gesundheitssystem, das oft versagt hat Bereitstellung der grundlegendsten präventiven und therapeutischen Versorgung für Erkrankte. Manche Patienten mit Sichelzellenanämie haben Angst davor, sich einer medizinischen Behandlung zu unterziehen, die auf dem neuesten Stand der Biotechnologie ist.
Aber Ärzte, die jahrelang Patienten beim Leiden beobachtet haben, und viele Eltern, die miterlebt haben, wie ihre Kinder jahrelange Qualen ertragen mussten, sind froh über das, was vor ihnen liegt.
„Wir sind endlich an einem Punkt angelangt, an dem wir uns allgemein verfügbare Heilmittel für die Sichelzellenanämie vorstellen können“, sagte Dr. John Tisdale, Direktor der Abteilung für Zell- und Molekulartherapeutika am National Heart, Lung and Blood Institute und Mitglied des Beratungsausschusses .
Dana Jones aus San Antonio möchte, dass ihre Töchter Kyra (18) und Kami (20) eine Chance auf eine der neuen Therapien bekommen. Beide hatten Schlaganfälle Dadurch erlitten sie Lernschwierigkeiten – Verletzungen, die wahrscheinlich hätten vermieden werden können, wenn sie einen Screening-Test und eine seit langem bekannte Behandlung erhalten hätten um neun von zehn Schlaganfällen zu verhindern bei Kindern mit dieser Krankheit. Kyra liegt jetzt auf der Intensivstation, während die Ärzte versuchen, ihre Schmerzen unter Kontrolle zu bringen.
Frau Jones ist überwältigt von der Möglichkeit, dass ihre Töchter geheilt werden könnten.
„Ich bete dafür, dass Kami und Kyra von dieser schrecklichen Krankheit geheilt werden und endlich wirklich leben können“, sagte sie.
Eine neue Behandlung und eine neue Technologie
Die Ursache der Sichelzellenanämie ist seit fast 70 Jahren bekannt, aber die Forschung hinkt hinterher. Viele sagen, dass die Situation zumindest teilweise darauf zurückzuführen ist, dass so viele Patienten Schwarze waren und aus armen Familien und der Arbeiterklasse stammten.
Es gibt eine Reihe von Behandlungen, um die Auswirkungen der Sichelzellenanämie zu reduzieren. Einige Patienten können eine Knochenmarktransplantation erhalten, die die Krankheit heilen kann. Dazu muss jedoch ein Spender gefunden und nach der Transplantation Medikamente eingenommen werden, um zu verhindern, dass der Körper die fremden Zellen abstößt.
In den letzten Jahren haben eine Reihe von Biotechnologieunternehmen neuartige Ansätze ausprobiert. Während Bluebird Bio seine Gentherapietechnik weiterentwickelt, konzentrierten sich Vertex und CRISPR Therapeutics auf das Gen-Editing-System CRISPR-Cas9, das bestimmte Bereiche der DNA angreifen und Gene ein- oder ausschalten kann. CRISPR hat es Forschern ermöglicht, Gene zu deaktivieren, um ihre Bedeutung für die biomedizinische Forschung zu bewerten. Bisher wurde es jedoch nicht zur Behandlung von Patienten mit einer genetischen Erkrankung eingesetzt.
Zur Behandlung von Sichelzellenanämie schneidet CRISPR ein Stück DNA in Knochenmarksstammzellen ab. Dadurch wird ein blockiertes Gen freigesetzt, um eine Form von Hämoglobin herzustellen, die normalerweise nur von einem Fötus produziert wird. Das fetale Gen steuert die Produktion von Hämoglobin, das keine Sichelform annimmt. In klinischen Studien litten die Patienten nicht mehr unter den Komplikationen der Sichelzellenanämie und benötigten keine Bluttransfusionen mehr.
Es besteht jedoch die Sorge, dass CRISPR versehentlich ein DNA-Stück im falschen Teil des Genoms eines Patienten herausschneiden könnte. Das könnte ein Gen stören und Blutkrebs verursachen.
In den klinischen Studien sind keine derartigen Probleme aufgetreten, aber an der Vertex-Studie waren nur 44 Patienten beteiligt, und nur 30 wurden mindestens 16 Monate lang beobachtet. Das Unternehmen führte umfangreiche Vergleiche der DNA von Patienten mit denen von Personen in großen Datenbanken durch und fragte, wie wahrscheinlich solche CRISPR-Aussetzer sein könnten.
Vertex sagte, es plane, Patienten in klinischen Studien 15 Jahre lang zu begleiten. Die Daten des Unternehmens waren so beruhigend, dass das Expertengremium am Dienstag erklärte, es sehe keinen Grund, die Behandlung zurückzuhalten.
Es kann immer noch zusätzliche Studien geben, bemerkte Ausschussmitglied Alexis Komor, Professor für Chemie und Biochemie an der University of California in San Diego. Aber, sagte sie, das würde bedeuten, „Perfektion auf Kosten des Fortschritts zu erwarten“.
Dr. Joseph Wu aus Stanford fügte hinzu: „Wir sind uns alle einig, dass die Vorteile die Risiken überwiegen. Diese Patienten sind ziemlich krank und das ist eine gute Therapie.“
Scot Wolfe von der Chan Medical School der University of Massachusetts sagte: „Wir wollen darauf achten, dass das Perfekte nicht der Feind des Guten ist.“
„Es besteht ein großer ungedeckter Bedarf“, fügte er hinzu.
Wenn es sicher ist, wer bekommt es?
Vertex schätzt, dass 20.000 Menschen Anspruch auf die Behandlung haben könnten, und sagt, Medicaid und private Versicherer hätten ihre Bereitschaft signalisiert, dafür zu zahlen.
„Es gibt fast keine Möglichkeit, wie sie das könnten nicht zahlen“, sagte Dr. David Williams, Leiter der Abteilung für Hämatologie und Onkologie am Boston Children’s Hospital.
Dr. Williams, der für Vertex und Bluebird Bio beratend tätig war, fügte hinzu, dass Versicherer „3 Millionen US-Dollar pro Stück“ für andere von Bluebird Bio hergestellte Gentherapien gegen die Krankheiten Thalassämie und Adrenoleukodystrophie zahlen. Bei Sichelzellenanämie und der großen Zahl schwarzer Patienten gebe es ein Problem mit der „Gleichberechtigung beim Zugang und dem enormen medizinischen Bedarf“.
Abhängig von den Entscheidungen der FDA sind einige Menschen mit dieser Krankheit möglicherweise nicht berechtigt. Dazu könnten kleine Kinder mit Sichelzellenanämie und ältere Patienten gehören, deren Körper so geschädigt ist, dass die Behandlung ein erhöhtes Risiko darstellen könnte.
Kevin Wake aus Kansas City, Missouri, hofft, dass er mit seinen 55 Jahren nicht zu alt oder zu geschädigt ist. Er hatte drei Schläge durch die Krankheit verursacht.
Die Behandlungen sind zwar heilend, aber schwierig.
Die Patienten erhalten zunächst acht Wochen lang Bluttransfusionen, gefolgt von einer Behandlung, um Knochenmarkstammzellen in ihren Blutkreislauf freizusetzen. Anschließend werden die Stammzellen entnommen und zur Behandlung an die Unternehmen geschickt. Anschließend erhalten die Patienten eine intensive Chemotherapie, um ihr Knochenmark von den behandelten Zellen zu befreien. Die behandelten Zellen werden den Patienten wieder infundiert, sie müssen jedoch mindestens einen Monat im Krankenhaus bleiben, während die neuen Zellen wachsen und ihr Knochenmark neu besiedeln.
Diese Behandlung „kann in den meisten Krankenhäusern nicht durchgeführt werden“, sagte Dr. Alexis Thompson, Leiterin der Abteilung für Hämatologie am Children’s Hospital of Philadelphia, die für Vertex berät.
Ein weiteres Problem ist, wie schnell Vertex die Produktion hochfahren kann. Die Zellen jedes Patienten müssen einzeln in einer sterilen Umgebung behandelt werden, eine mühsame Angelegenheit.
Stuart Arbuckle, Executive Vice President und Chief Operating Officer bei Vertex, ist zuversichtlich. „Wir sind startbereit“, sagte er. Er fügte jedoch hinzu, dass er nicht sofort mit einer großen Patientenwelle rechnet.
„Das ist eine ziemlich große Entscheidung für einen Patienten“, sagte Herr Arbuckle.
Eine der Patienten der klinischen Vertex-Studie, Marie-Chantal Tornyenu, 22, Studentin an der Cornell University, sagte, dass die Patienten auch auf eine „mentale Anpassung“ nach der Behandlung vorbereitet werden müssten.
Frau Tornyenu sagte, sie habe nicht mehr die Schmerzkrisen gehabt, die sie geplagt hätten, insbesondere in der Highschool, als sie fast jeden Monat ins Krankenhaus musste.
Aber sie hat einen Großteil ihres Lebens damit verbracht, Vorsichtsmaßnahmen zu treffen und sich Sorgen über Schmerzen und Komplikationen durch Sichelzellenanämie zu machen. Diese Gewohnheiten sind schwer zu durchbrechen.
„Es ist eine große Lernkurve, weil ich mein ganzes Leben lang Sichelzellenanämie hatte“, sagte sie. „Ich kämpfe immer noch mit dieser Denkweise – ‚Sichelzellenanämie bist du‘.“
[ad_2]